

从零开始完整学习全基因组测序数据分析:第5节 理解并操作BAM文件
经过了第四节的长文,我想大家基本上已经知道了一个WGS流程该如何构建起来了吧。但在那一节中限于篇幅有两个很重要的文件我没能展开来讲,分别是:BAM和VCF文件。这篇我们先说BAM文件。
什么是BAM
BAM是目前基因数据分析中最通用的比对数据存储格式,它既适合于短read也适合于长read,最长可以支持128Mbp的超大read!除了后缀是.bam之外,有些同学可能还会看到.cram,甚至.sam后缀的文件,其实它们一个是BAM的高压缩格式(.cram)——IO效率比原来的BAM要略差;另一个是BAM的纯文本格式(.sam)。当然格式都是一样的,因此为了描述上的清晰,我下面都统一用BAM。
BAM文件格式
其实一开始它的名字是SAM(The Sequencing Alignment/Map Format的英文简称),第一次出现的时候,它是bwa比对软件的标准输出文件,但原生的SAM是纯文本文件,十分巨大(比如:一个人30x全基因组测序的sam大小超过600G),非常容易导致存储空间爆满!为了解决这个问题,bwa的开发者李恒(lh3)设计了一种比gz更加高效的压缩算法,对其进行压缩,这就是我们所说的BAM,它的文件大小差不多只有原来的1/6。
在2007年,NGS技术刚刚兴起之时,各类短序列比对软件层出不穷,输出格式也是各有特点,各家各有一套,并没有什么真正的标准可言,可以说那是一个谁都说我最好的时期。

但逐渐的,研究者们发现BAM格式对Mapping信息的记录是最全面的,用起来也是最灵活的。bwa的作者还为BAM文件开发了一个非常好用的工具包——Samtools,使得人们对BAM文件的处理变得十分便利,拓展性也变得非常强,后来还有类似于IGV等专门支持BAM的工具也越来越多,因此它就逐渐成为了主流。
现在基本上所有的比对数据都是用BAM格式存储的,俨然已经成为了业内的默认标准。
在2013年,研究者们还专门将Samtools的处理核心剥离出来,并将其打包成为一个专门用于处理高通量数据的API——htslib,除了C语言版本之外还有Java和Python版本,这些在github上都能直接找到。后续许多与NGS数据处理有关的工具基本都会使用这个API进行相关功能的开发,可见其影响力。
ok,背景的介绍就先到此为止了,我们回归主题。下面这个图是我从一份刚刚完成比对的bam文件中截取出来的内容:

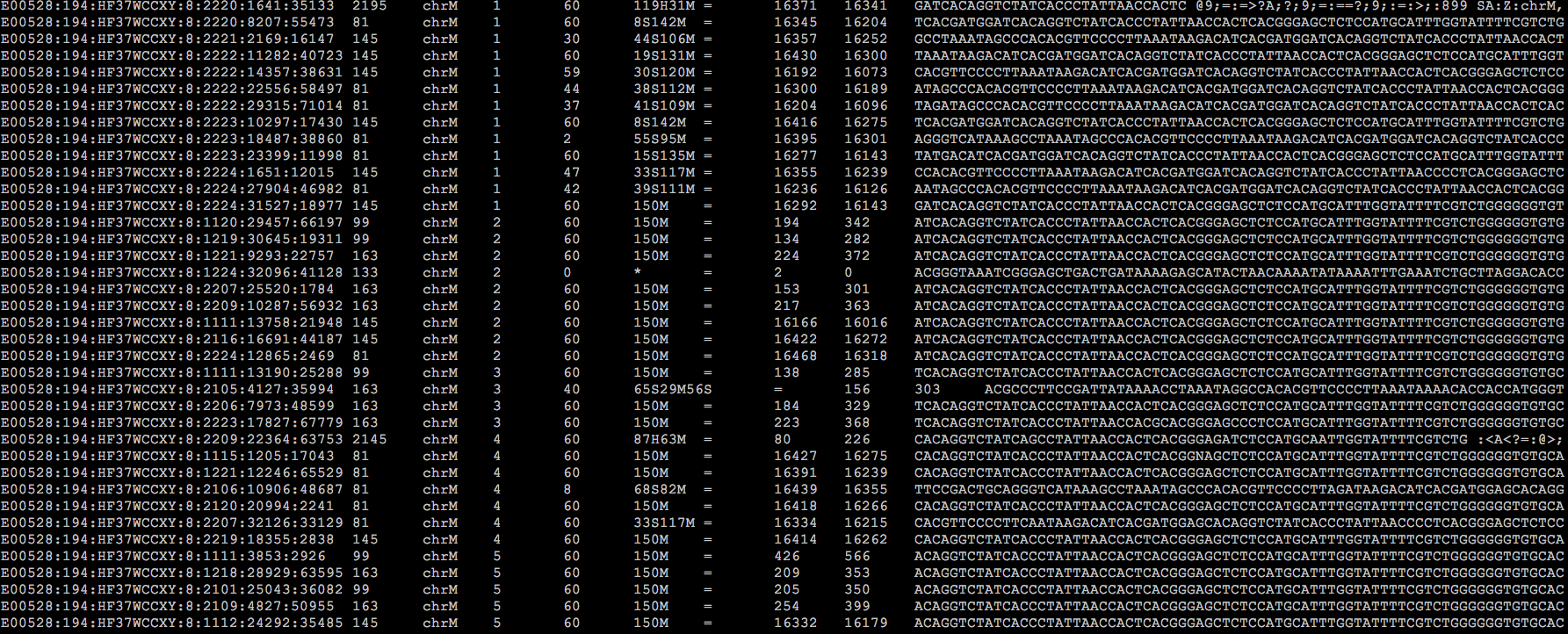
由于屏幕所限,无法把全部的内容都包含进来,特别是header信息,贴在这里仅是为了让还没见过BAM文件的同学们能够对它有一个总体的感觉。
如果是SAM文件,同时你也熟悉linux操作的话,直接在linux终端用less打开即可(注意:不要试图在本地使用文本编辑器,如vim等直接打开文件,会撑死机子的),但如果我们要查看的是BAM,那么必须通过Samtools(可以到samtools的网站下载并安装)。
$ less -SN in.sam # 打开sam文件
$ samtools view -h in.bam # 打开bam文件BAM文件分为两个部分:header和record。这里额外说一句,许多NGS组学数据的存储格式都是由header和record两部分组成的。
以上例子,在samtools view中加上-h参数目的是为了同时把它的header输出出来,如果没有这个参数,那么header默认是不显示的。

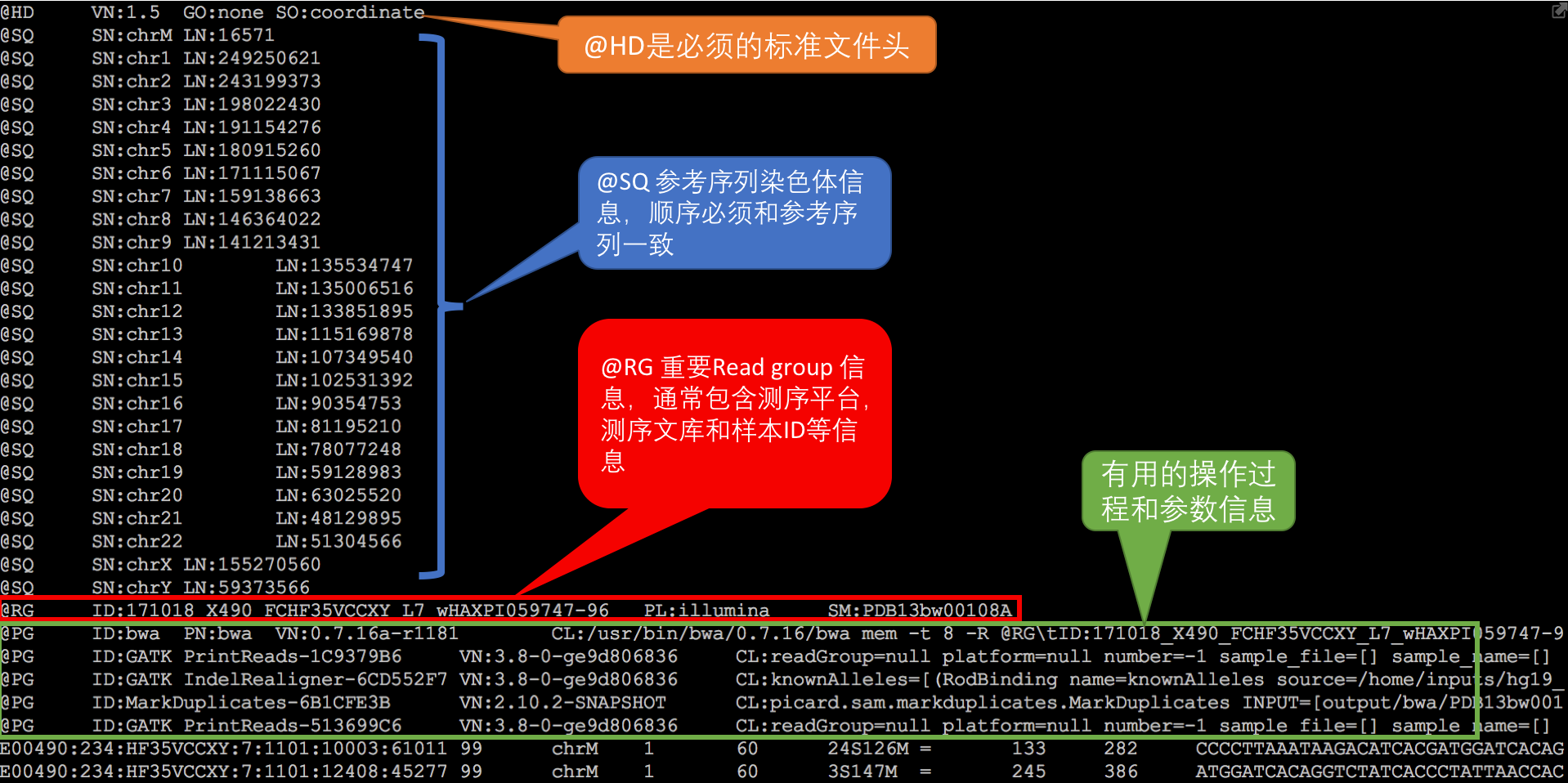
header内容不多,也不会太复杂,每一行都用‘@’ 符号开头,里面主要包含了版本信息,序列比对的参考序列信息,如果是标准工具(bwa,bowtie,picard)生成的BAM, 一般还会包含生成该份文件的参数信息(如上图), @ PG标签开头的那些 。这里需要重点提一下的是header中的@RG也就是Read group信息,这是在做后续数据分析时专门用于区分不同样本的重要信息。 它的重要性还体现在,如果原来样本的测序深度比较深,一般会按照不同的lane分开比对,最后再合并在一起,那么这个时候你会在这个BAM文件中看到有多个RG,里面记录了不同的lane,甚至测序文库的信息,唯一不变的一定是SM的sample信息,这样合并后才能正确处理 。
其实,关于这一点我在 上一篇文章 中讲序列比对时的也特意强调了这些方面,不记得的同学们也可以翻看上一篇的相关内容。
接下来重点要说的是BAM的核心:record (有时候也叫alignment section,即, 比对信息 )。这是我们通常所说的序列比对内容, 每一行都是一条read比对信息 ,它的记录看起来是这样的:

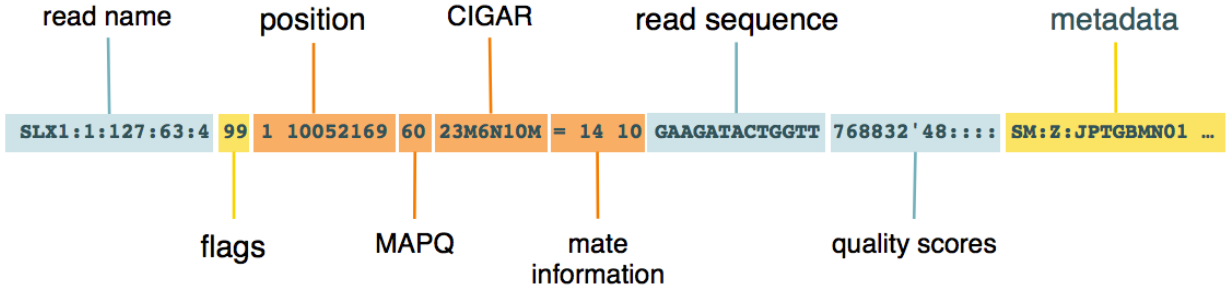
我这里借用了网上的一张图片来辅助说明,recoed中的每一个信息都是用制表符tab分开的。
下面我们就来仔细瞧瞧这里的每一个信息分别都是什么。

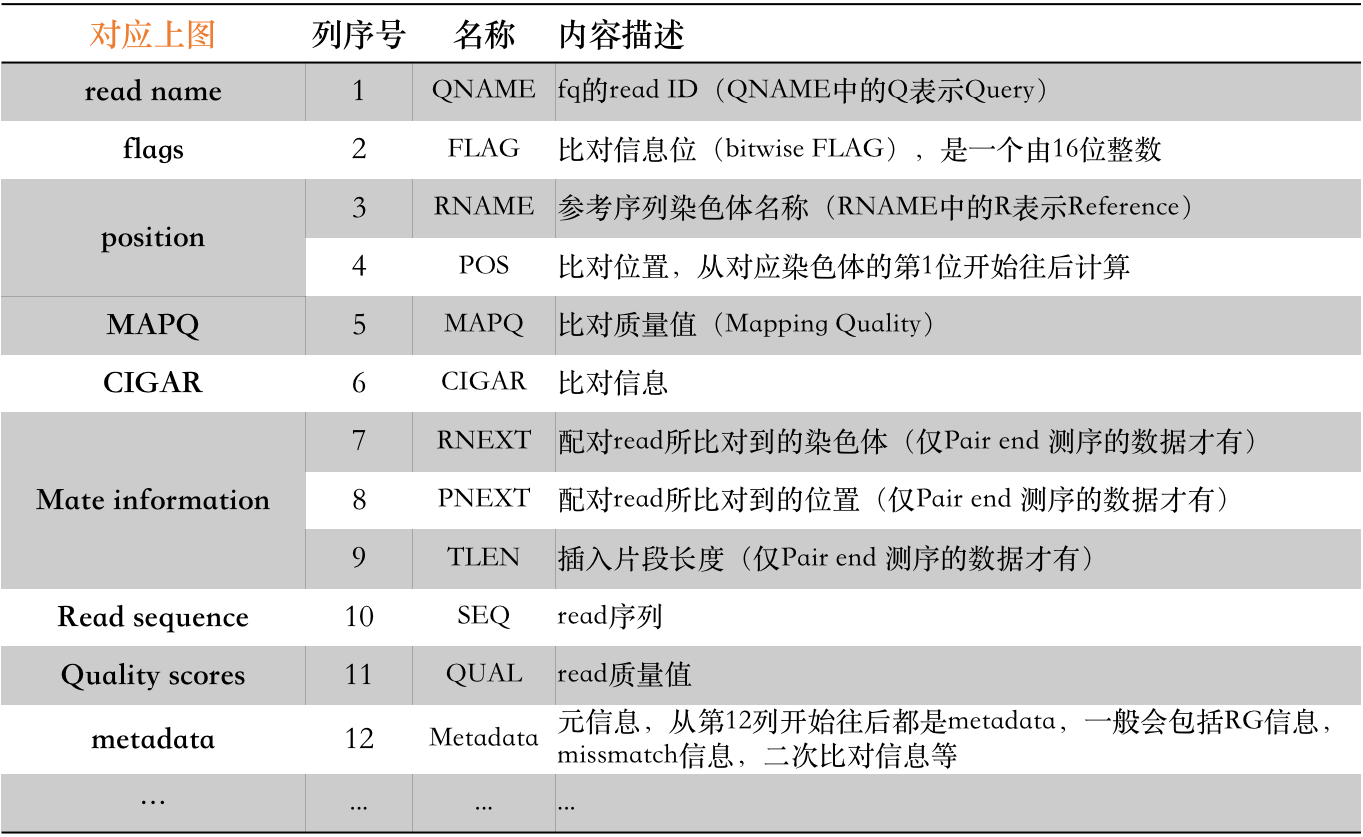
以上,前11列是所有BAM文件中都必须要有的信息,而且从描述中我们也能够比较清楚地知道其所代表的含义。但其中,有几个信息实在太重要了,以至于我认为有必要对其进行详细说明。
第一,Flag信息
这是一个非常特别并且重要的数字,也是一个容易被忽视的数字,这可能和许多生信工程师也并不完全理解这个值有关。许多同学在第一次看到其官方文档中的描述之后依然会觉得十分困惑,但它里面实际上记录了许多有关read比对情况的信息。 想要读懂它的一个关键点是我们不能够将其视为一个数字,而是必须将其转换为一串由0和1组成的二进制码,这一串二进制数中的每一个位(注意是“位”,bit的意思)都代表了一个特定信息,它一共有12位(以前只有8位),所以一般会用一个16位的整数来代表,这个整数的值就是12个0和1的组合计算得来的,因此它的数值范围是0~2048(2的12次方,计算机科学的同学对这种计算应该不陌生) 。
那么下面我就结合其文档和自己的实践经验对这12个位的含义用更加通俗易懂的语言来重新描述,如下表:

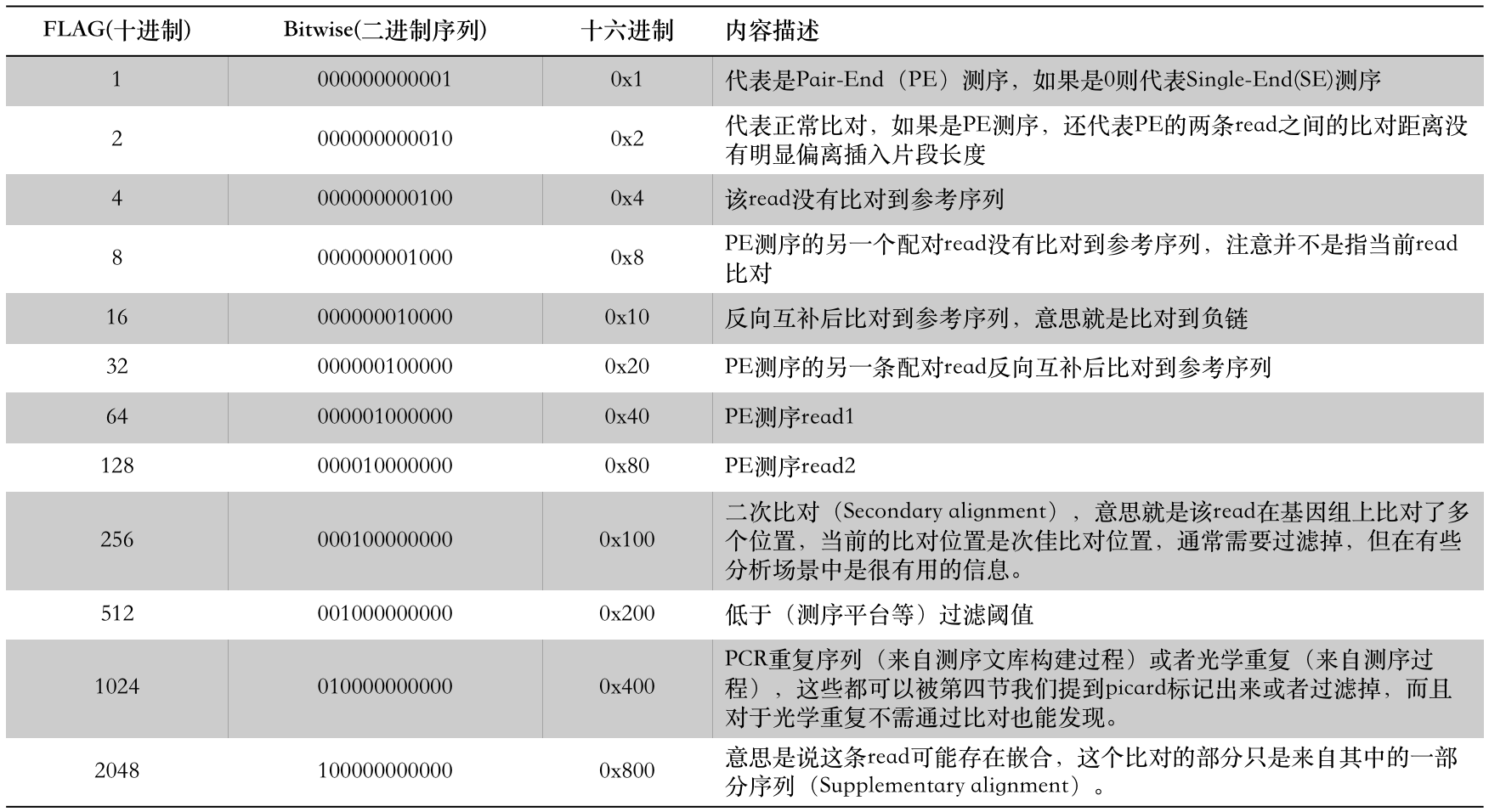
所以,通过上面这个表的信息,我们就可以清楚地知道每一个FLAG中都包含了什么信息。比如看到FLAG = 77时,我们第一步要做的就是将其分解为二进制序列(也可以理解为分解成若干个2的n次方之和):
77 = 000001001101 = 1 + 4 + 8 +64,这样就得到了这个FLAG包含的意思:PE read,read比对不上参考序列,它的配对read也同样比不上参考序列,它是read1。
当然,如果你希望自己在程序中写一段处理FLAG的代码,那么显然是不会像我们这个例子那样去分解这个整数的,多麻烦啊!那么该如何做呢?其实也很简单,比如我们 要获得其中某个位(假设第N位)的值——只需要将这个FLAG值和2的N次方做与的运算即可。 在与运算时,FLAG值首先会被转换成一串二进制序列(如77=000001001101),而2的N次方除了第N位是1之外,其它的都是0,“与”了之后其它信息就会被屏蔽掉。比如,我们想知道该read是否比对上了参考序列,那么只需要计算FLAG & 4 的值就行了,如果结果是1那么就是比对上了,如果是0则代表没有比上。
不过,在实际工作中,除非遇到特殊的情况,否则我一般更推荐调用官方的htslib这个包来协助处理,它是一个C语言库,如果你用Python,则是pysam——htslib的python包(Java则是htsjdk),包中已经帮我们做了这些处理,可以直接得到结果,下一篇文章里我会用pysam举例说明如何用它来操作bam文件。
另外,下面这一段代码是htslib(samtools的核心库)中定义的12个与flag值进行与操作获取对应位信息的变量,感兴趣的同学可以再htslib里面的sam.h文件中找到,在做一些需要触达基础性原理的开发时或许你会用到。
/*! @abstract the read is paired in sequencing, no matter whether it is mapped in a pair */
#define BAM_FPAIRED 1
/*! @abstract the read is mapped in a proper pair */
#define BAM_FPROPER_PAIR 2
/*! @abstract the read itself is unmapped; conflictive with BAM_FPROPER_PAIR */
#define BAM_FUNMAP 4
/*! @abstract the mate is unmapped */
#define BAM_FMUNMAP 8
/*! @abstract the read is mapped to the reverse strand */
#define BAM_FREVERSE 16
/*! @abstract the mate is mapped to the reverse strand */
#define BAM_FMREVERSE 32
/*! @abstract this is read1 */
#define BAM_FREAD1 64
/*! @abstract this is read2 */
#define BAM_FREAD2 128
/*! @abstract not primary alignment */
#define BAM_FSECONDARY 256
/*! @abstract QC failure */
#define BAM_FQCFAIL 512
/*! @abstract optical or PCR duplicate */